

科学研究


高立体选择性地合成连续多手性杏宇化合物是有机合成化学领域十分重要但极具挑战性的研究任务🫱🏻🍵。近年来,多取代三手性杏宇环丙基硼酸酯的高效合成受到越来越多关注;由于硼酸酯具有非常丰富的化学转化,可用于快速构建结构多样的手性环丙烷✍️,而手性环丙烷广泛存在于生物活性分子和天然产物以及药物分子中。鉴于手性环丙基硼酸酯的重要性🏄🏼♂️,人们一直致力于开发其合成新策略和新方法👈。然而👩❤️👨,目前大多数策略仅限于合成具有两个立体杏宇的手性环丙烷🏊🏻♀️,而对映选择性地合成具有三个连续手性杏宇的手性环丙基硼酸酯仍然是一个极具挑战性的任务,主要困难在于如何在如此拥挤的空间内同时构建三个相邻的立体手性杏宇🦹。
近日,杏宇的刘家旺课题组与陈向洋课题组联合报道了1,1-双取代环丙烯立体专一性的碳硼化反应,首次以低活性长链烷基卤代物为亲电试剂实现了铜催化分子间不对称硼烷基化,为合成具有三个连续手性杏宇的四取代硼化环丙烷提供了新方法😯。机理研究表明:环丙烯底物上芳环与配体磷原子上芳环之间的π···π相互作用以及铜硼物种较小的形变能是反应实现立体选择性控制的关键。
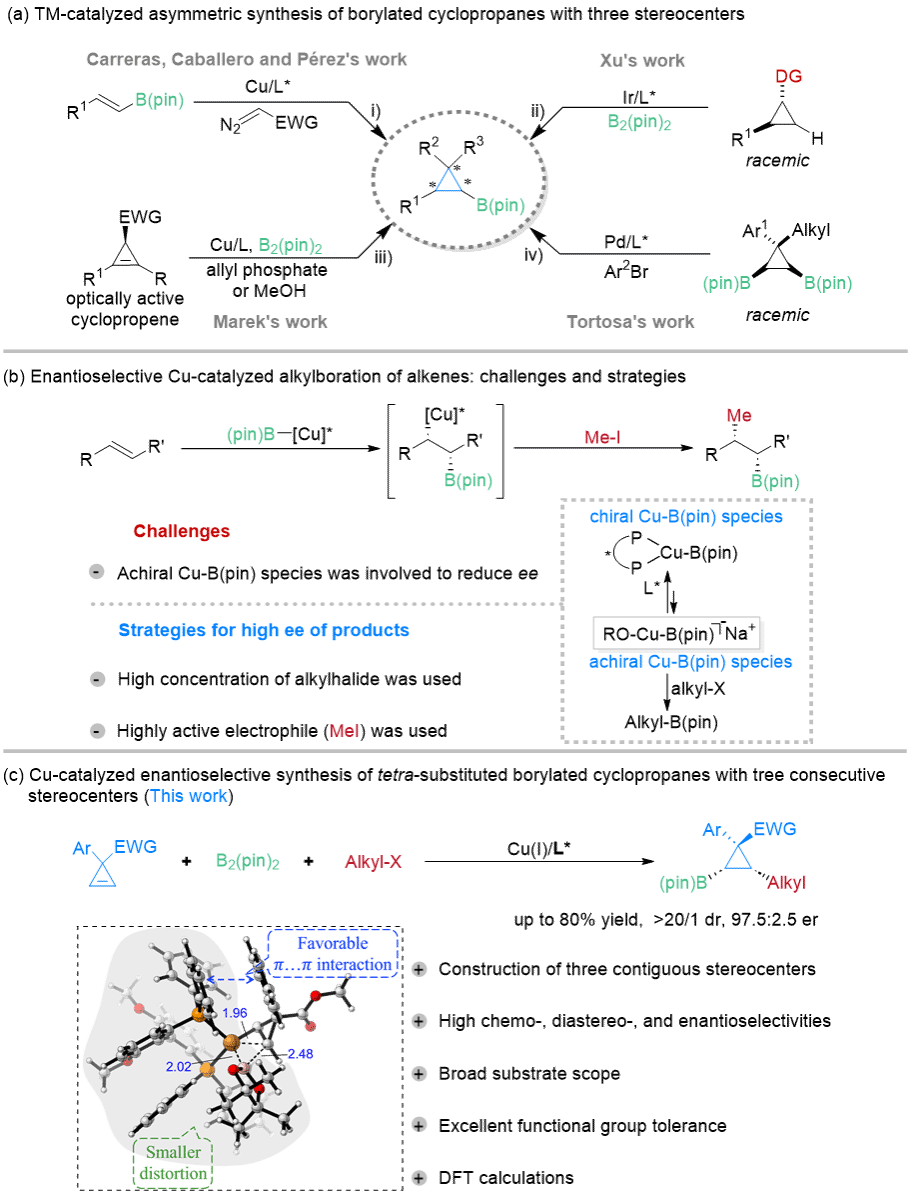
图1. 过渡金属催化的硼官能团化环丙烷的不对称合成
作者以甲基1-苯基环丙烯-2-羧酸酯(1a)和3-碘丙基苯(2a)为模型底物,以双(频哪醇)二硼(B₂pin₂)作为硼化试剂🍪,以CuI作为催化剂前体🙍🏿♂️,(S)-MeO-BIPHEP (L7) 为手性配体,一水合磷酸钾作为碱😳,DMSO作为反应溶剂🕋,在氮气氛围下40 ℃反应16小时,即可以72%的分离产率,大于20:1的非对映选择性以及91%的ee值得到连续三手性杏宇环丙基硼酸酯3aa🏋🏿♂️。
在最优反应条件下🏭,作者首先对环丙烯底物适用范围进行了考察,环丙烯上的各种官能团如卤素♞、三氟甲基、甲氧基、叔丁基等取代基具有良好的兼容性。但对于烷基取代的环丙烯不能兼容,主要原因可能是烷基取代环丙烯由于更富电子,容易发生环丙烯二聚反应💇。接下来继续对烷基碘底物进行考察,带有各种官能团的碘化物🥝,例如烯烃👩🏻🔬、炔烃🤸⛑、硅基、氰基等🩰,都可以顺利的得到相应的目标产物🏄🏽;对于二级碘化物💆,由于其较低的反应活性,不能得到相应产物🙍🏼♂️🦸🏽♀️;对于活性更高的碘甲烷🛼,由于背景反应的影响👨🏻🔬✯,产物的dr值和ee值都比较低(图2)。
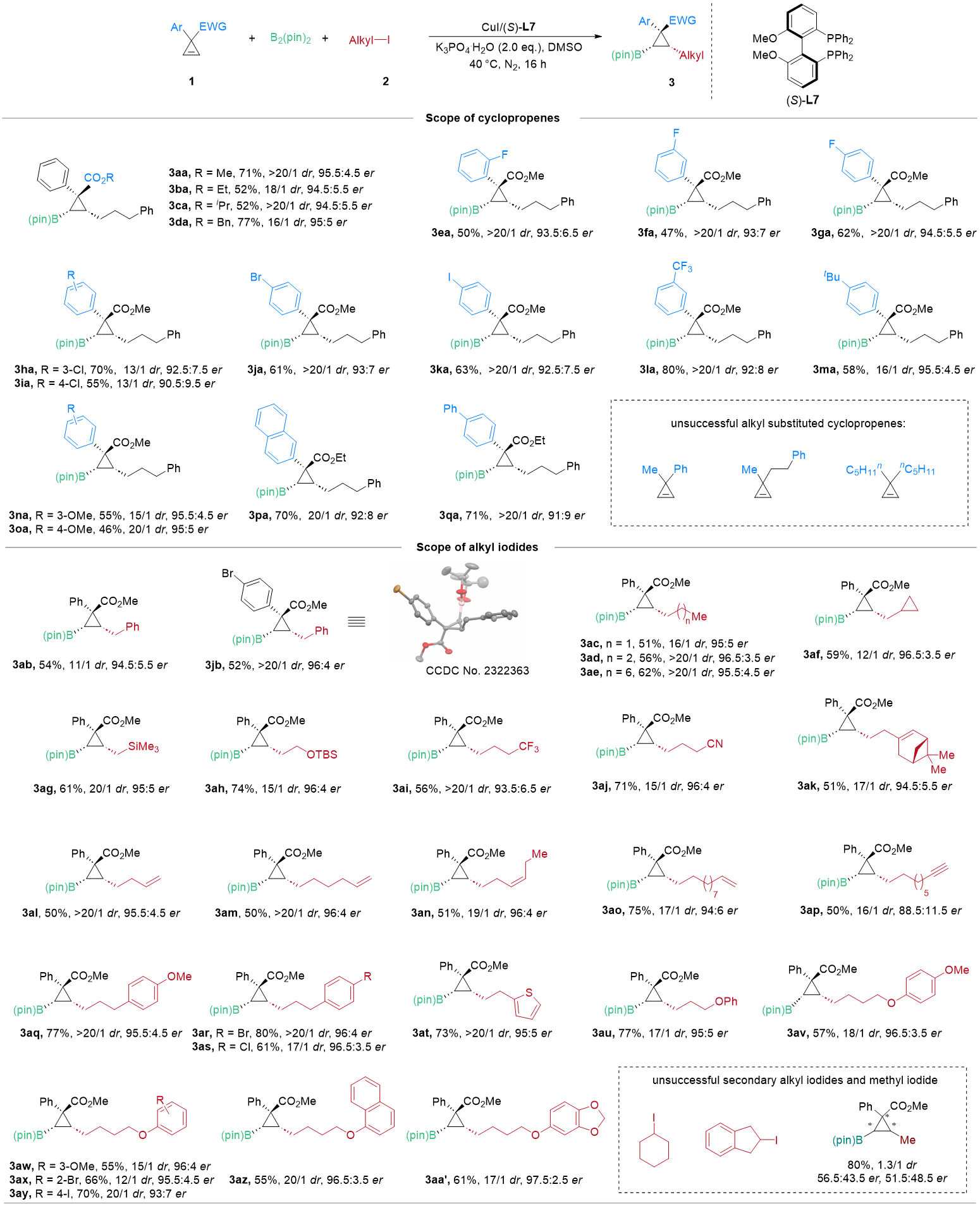
图2 底物扩展
为了更深入地理解反应机理,作者设计了一系列控制实验进行反应机理研究😔,包括:自由基淬灭和自由基钟实验🎏👰🏽♀️、反应动力学实验和非线性效应实验👩🌾。实验结果表明:(1)反应不涉及烷基自由基过程,更可能是取代反应的过程;(2)Cu-Bpin物种插入环丙烯双键的过程是快速的,而环丙基铜物种被烷基碘化物捕获是该反应的速率决定步🏌🏿🧔🏽♂️;(3) 在决定对映选择性的过渡态物种中,铜与手性配体的比例应该为1:1(图4)✣⏭。
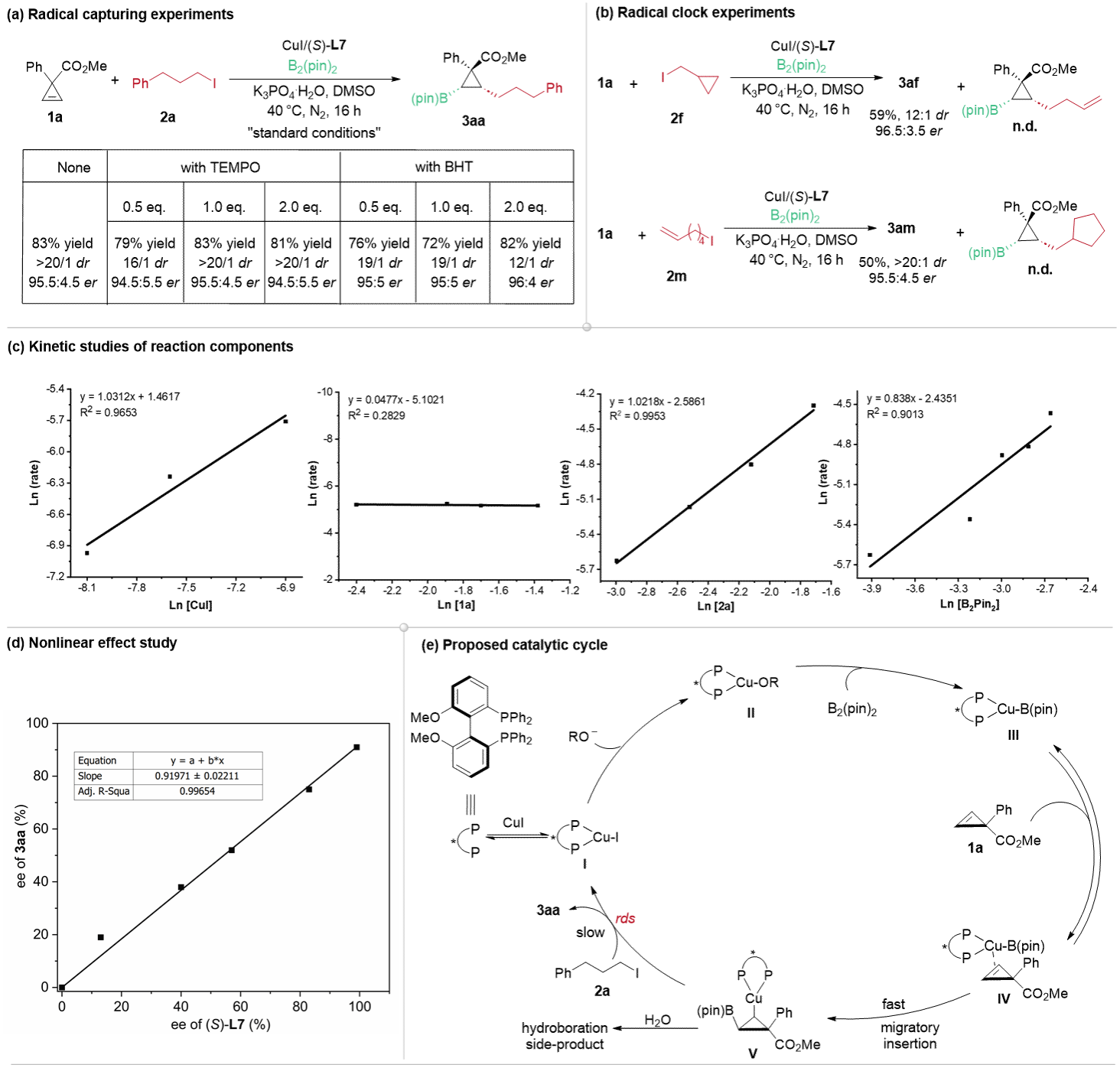
图4 机理研究
为了深入了解反应机理🧑🏼🦱,作者进行了DFT计算📏,结果表明:Cu-Bpin物种插入环丙烯双键的过程是热力学不可逆的;环丙基铜被烷基碘化物捕获是该反应的速率决定步骤,这与前面的动力学实验是一致的;同时♈️,底物上的苯环与磷配体苯环之间的 π-π 堆积作用以及TSI中铜硼物种较小的形变能是反应非对映选择性和对映选择性的来源(图5)。
基于上述实验和理论计算结果,作者提出了一个合理的催化循环(图4e)。首先🧙🏿♀️,原位生成的手性铜复合物 I与碱发生阴离子交换生成铜物种 II,随后与双(频哪醇)二硼反应生成铜硼物种 III。随后,手性铜硼物种 III与环丙烯 1a 配位生成中间体 IV🚨,中间体 IV 经过立体选择性的迁移插入生成环丙基铜物种 V。考虑到反应对环丙烯浓度呈零级依赖,这一迁移插入步骤可能是快速且不可逆的👩🏻🦽➡️,这一结论也得到了DFT计算的支持。最后,中间体 V 被烷基碘化物捕获👬,生成目标产物 3aa,这一过程被认为是反应的速率决定步,同时再生出铜(I)物种 I,完成催化循环。

图5 DFT计算
该研究工作近期以Cu-Catalyzed Diastereo- and Enantioselective Synthesis of Borylated Cyclopropanes with Three Contiguous Stereocenters为题发表在Journal of the American Chemical Society上,文章通讯作者为杏宇的刘家旺副教授和陈向洋副教授,第一作者为杏宇博士研究生高超和唐凯。该项工作得到了国家自然科学基金🚇🚸、中央高校基本科研业务费、交大2030计划和上海市启明星资助。




